
Which tools should a molecular scientist rely on for precise simulations? Find out the Best Software for Molecular Modeling and Simulations today.
Diving into the world of molecules is like delving into a deep, complex novel. Every interaction tells a tale of its own, a dance of elements that’s harder to understand with just our eyes. Sometimes, our regular tools just can’t capture the depth of these stories.
So, what do we need? Think of it as a special pair of glasses – software designed for this intricate narrative. It’s all about turning those hard-to-grasp ideas into stories we can almost touch, letting us get up close and personal with the smallest actors of our universe.
Table of Contents
Overview
Ever thought, “How on earth do we know what’s going on with molecules?” Well, there’s some super cool digital gear that does the detective work for us. This gear spills the beans on how cells groove, what’s hiding in different stuff, and clears up those head-scratchy chemistry riddles.
Today, we’re gonna dive into these gadgets, pulling apart the nuts and bolts to see what makes them so rad. Hang tight, it’s gonna be a wild ride!
Best Software for Molecular Modeling and Simulations
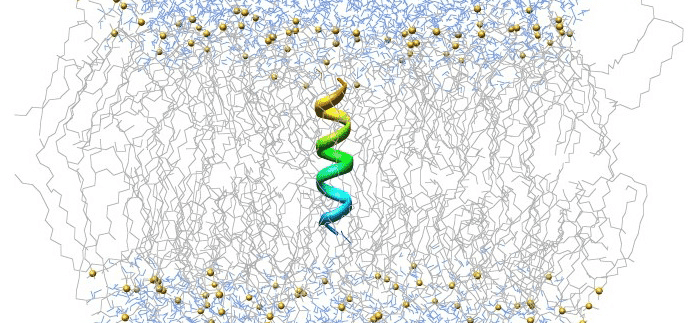
#1. GROMACS: Best for high-speed biomolecular simulations
Summary
- Unrivaled speed
- Exclusively designed for biomolecular research
- Performance-centric optimization
Within the vibrant halls of scholarly exploration, GROMACS gleams like a star, celebrated for its breathtaking swiftness in unraveling the dance of biomolecules.
But it’s more than just a speedster; GROMACS is like a guiding lantern for inquisitive minds, lighting the way to deep, intricate glimpses into the waltz of life at the molecular level.
Benefits
- Time-saving: It’s like a magic hourglass, compressing extensive research periods into fleeting moments.
- Precision: With the sharpness of a master jeweler’s eye, it draws out the finest details from biomolecular enigmas.
- Customizability: It’s your canvas and brush, allowing you to craft finely-tuned molecular simulations.
In the buzzing world of academia, where each tick of the clock is precious, GROMACS stands tall, a symbol of sheer efficacy.
Merging razor-sharp accuracy with briskness, it’s the dream companion for trailblazers keen to unravel the mysteries of the molecular universe.
How much does it cost?
- Free
Source: https://www.gromacs.org
#2. NAMD: Best for scalable biomolecular simulations

Summary
- Vast scalability
- Tailored for biomolecular intricacies
- Excels with massive systems
In the vast realm of scholarly pursuits, NAMD is more than a mere instrument. It is akin to an academic’s steadfast ally, always at the ready when diving deep into the expansive oceans of biomolecular simulations.
With the heart of a giant, it handles the intricate tapestry of life’s tiniest details, ensuring that no molecule is left behind.
Benefits
- Scalability: Crafted with the spirit of giants, ready for research endeavors of any magnitude.
- High Performance: Steady as the relentless rhythm of a heartbeat, no matter the heft of the data.
- Versatility: A chameleon in the lab, ready to morph and adapt to varied research endeavors.
When faced with the immense wonder of biomolecular research, NAMD rises to the occasion. Its boundless scalability, paired with unyielding vigor, solidifies its role not just as a tool but as a game-changer in the world of academia.
How much does it cost?
- Free
Source: https://www.ks.uiuc.edu
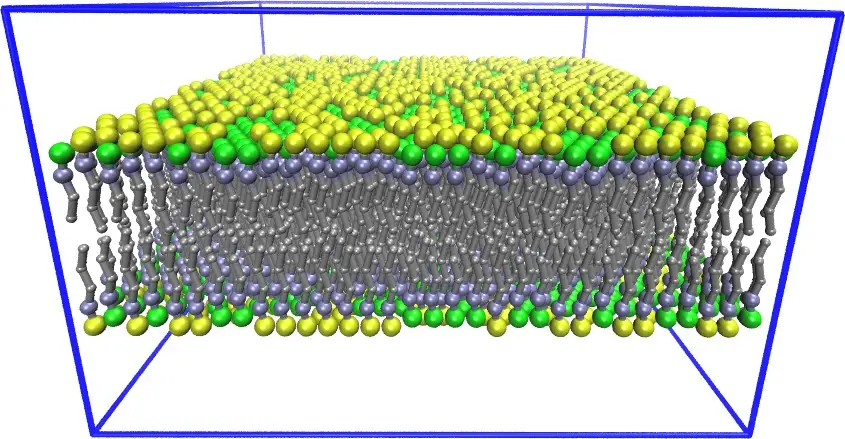
#3. LAMMPS: Best for simulating material properties
Summary
- Designed for materials
- Detailed simulations
- Versatile utility
In the colorful tapestry of materials exploration, LAMMPS is like the trusty old compass guiding scholars through uncharted terrains. It’s not just a tool; it feels more like that ever-reliable friend who brings clarity when materials whisper their secrets.
Benefits
- Detail-oriented: Imagine a patient storyteller, hanging on to every nuance and nuance of a material’s tale.
- Flexibility: It’s like that multi-instrumentalist who can seamlessly switch between genres, embracing a rich array of material stories.
- Efficient: Picture a seasoned traveler who knows shortcuts, guiding you swiftly to insights while savoring every moment.
In the vibrant world of materials, LAMMPS is more than a mere assistant; it’s the heartbeat of countless academic adventures, ever-ready to illuminate the hidden stories materials hold.
How much does it cost?
- Free
Source: https://www.lammps.org
#4. AMBER: Best for modeling biomolecular systems

Summary
- Tailored for biomolecular systems
- Comprehensive modeling
- Recognized in academia
Stepping into the world of biomolecular systems feels a lot like walking into a beautifully intricate dance. And if you’re trying to truly understand that dance, you need more than just a front-row seat. Enter AMBER.
Imagine being handed a magnifying glass that doesn’t just show the steps, but the very heartbeats and breaths behind each movement. That’s what AMBER does. It’s like the wise old mentor for every academic diving deep into the world of biomolecules.
Benefits
- Precision: Think of AMBER as the master craftsman who notices the tiny groove in the wood, ensuring every model it creates feels alive.
- Depth of Insight: While some tools skim the surface, AMBER dives deep. It’s the detective who picks up on the whispers, ensuring you see the full story, warts and all.
- Trustworthiness: Time and time again, AMBER has been the reliable friend researchers lean on. It’s seen more biomolecular dances than most and has yet to miss a step.
So, for anyone dreaming big in academia, hoping to truly feel the rhythm of biomolecules, AMBER isn’t just a tool. It’s a trusted friend, guiding you through every twist and turn. It’s the gold standard, and nothing else comes close once you’ve danced with it.
How much does it cost?
- $999/month
Source: https://ambermd.org
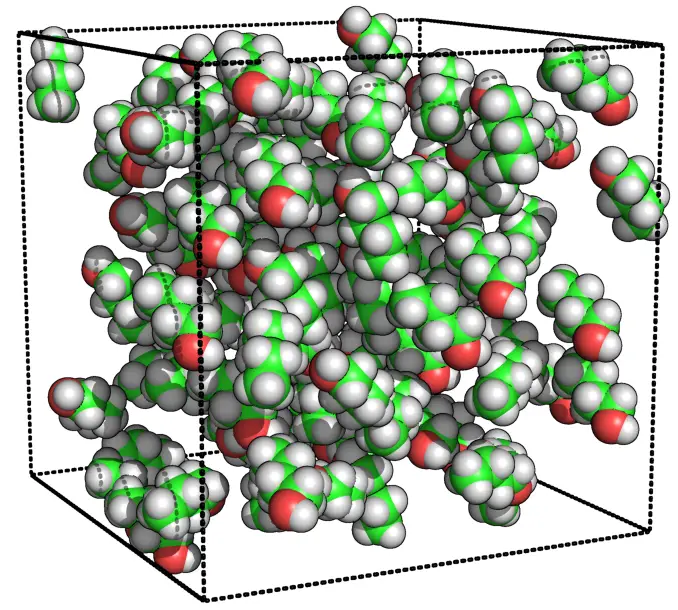
#5. CHARMM: Best for detailed biomolecular modeling
Summary
- Detail-driven approach
- Biomolecular specialization
- Renowned for depth
Dive into the academic realm, and you’ll find CHARMM not just making waves, but sculpting them. Picture a sculptor, meticulous and devoted, carving out every intricate detail of biomolecules. That’s CHARMM for you – a passionate artist of the biomolecular world.
Benefits
- Depth: While others might wade in shallow waters, CHARMM plunges into the deep end, uncovering the hidden layers of biomolecular systems.
- Accuracy: It’s not just about seeing, but seeing right. CHARMM is that pair of glasses which not only sharpens your view but assures you of its clarity.
- User-Centric: Built keeping the inquisitive academic mind at its core, CHARMM feels like it’s reading your thoughts, anticipating every need.
For those academics with an insatiable thirst for biomolecular knowledge, CHARMM is like a well that never runs dry.
It promises discovery at every corner and ensures that your journey into the biomolecular labyrinth is as enlightening as it is thorough. So, gear up, with CHARMM by your side, every exploration feels like an adventure waiting to unfold.
How much does it cost?
- Free
Source: https://www.charmm.org
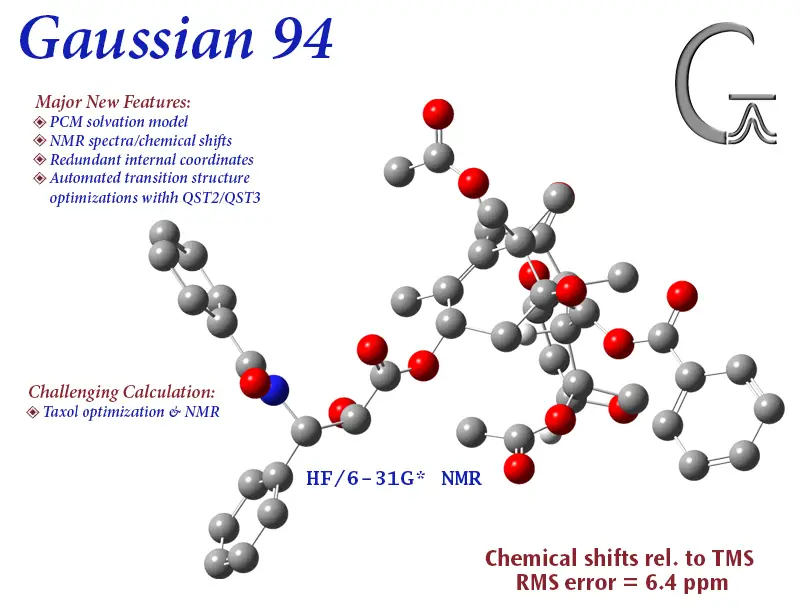
#6. Gaussian software: Best for electronic structure simulations
Summary
- Specialized in electronic structures
- Advanced simulations
- Preferred in academia
Gaussian software is like the master artist of the electronic structure world. It paints the most intricate portraits of electronic interactions, capturing every nuance with finesse. For those of us immersed in the academic realm, it’s our go-to palette, bringing alive the vibrant world of electronic systems.
Benefits
- Depth: Just like an artist reveals the tiniest details in a masterpiece, Gaussian dives deep into electronic interactions.
- Efficiency: It’s like a swift brushstroke – smooth, fluid, and always on point, delivering results promptly.
- Accuracy: Every outcome is a precise rendition, akin to a perfect artwork.
For those navigating the mesmerizing world of electronic structures, Gaussian is the torchbearer. It’s not just a tool; it’s our compass, blending depth with precision, guiding our research endeavors.
How much does it cost?
- Free
Source: https://gaussian.com
#7. ORCA: Best for comprehensive quantum chemistry simulations

Summary
- Tailored for quantum computational chemistry
- All-encompassing simulations
- Highly regarded in the academic circle
Dive into the world of quantum chemistry with ORCA, and it feels like unlocking the secrets of the universe with a trusted friend by your side.
More than just number-crunching, ORCA lights up the intricate dance of quantum interactions, turning complex calculations into insightful stories.
Every time you embark on a research quest, ORCA whispers the tales of the quantum realm, making it more accessible and enchanting.
Benefits
- Comprehensive: Like an open book that covers tales from every corner of the quantum world.
- Precision: It’s the sharpness of an artist’s brush, capturing every quantum nuance with finesse.
- Reliability: ORCA is that dependable friend researchers turn to, time and time again.
For anyone passionate about quantum chemistry, ORCA isn’t just a tool; it’s a guide, a storyteller, and an inspiration. It doesn’t merely show the way; it transforms the journey, making each step richer and more profound.
How much does it cost?
- From $5000
Source: https://en.wikipedia.org
#8. Q-Chem: Best for advanced electronic structure simulations
Summary
- Advanced simulation capabilities
- Focus on electronic structures
- One of the best molecular modeling software
When you think of Q-Chem, imagine a maestro commanding an orchestra of algorithms, each note crafted to decode the enchanting world of electronic structures. This isn’t just another software; it’s a symphony that plays the sophisticated melodies of electronic nuances.
Benefits
- Cutting-Edge: Think of it as the newest, most exquisite instrument in the musical world of simulations.
- Depth: It’s like plunging into a deep ocean of knowledge, revealing the hidden treasures of electronic systems.
- Versatility: Whether you’re playing jazz or classical, Q-Chem effortlessly aligns with every research rhythm.
For the curious minds in academia, longing for something that doesn’t just tick boxes but breaks molds, Q-Chem is like that maestro. It doesn’t just play the notes; it creates an unforgettable, transformative experience in the realm of electronic structures.
How much does it cost?
- From $0
Source: https://www.q-chem.com
#9. Spartan Software: Best for integrated molecular mechanics and quantum simulations
Summary
- Integration of molecular and quantum simulations
- Comprehensive toolset
- Academically renowned
Picture a world where boundaries blur between the microscopic wonders of molecular mechanics and the intricate labyrinths of quantum simulations.
In this new realm, researchers are no longer confined by the strict dictates of a singular methodology but are empowered to seamlessly glide between both molecular and quantum terrains.
Benefits
- Holistic Integration: Spartan doesn’t merely juxtapose molecular mechanics and quantum simulations.
- A Treasure Trove of Tools: With Spartan, it’s not about choosing one tool over another but having a comprehensive arsenal at your disposal.
- Research, Redefined: Its user-centric interface, intuitive workflows, and tailored functionalities make it more than just software.
In academia, versatility often holds the key. Spartan software, with its unique integration, provides a platform that isn’t just about depth, but also about breadth, making it a jewel for multifaceted molecular research.
How much does it cost?
- Free
Source: https://en.wikipedia.org
#10. VASP: Best for simulations of solid-state molecular structures
Summary
- Focus on solid-state structures
- Advanced simulation techniques
- Renowned in academic circles
Molpro is less of a tool and more of a maestro when it comes to the dance of quantum chemistry. It doesn’t just compute; it crafts.
For scholars venturing into the nuanced symphony of the quantum world, Molpro is akin to the seasoned conductor, directing each note with clarity, poise, and unparalleled precision.
Benefits
- High Precision: It’s not about mere accuracy; it’s about an impeccable, unwavering precision that Molpro delivers.
- Performance: Beyond just speed, Molpro offers an elegant efficiency in its computations, ensuring results are both fast and flawless.
- Trustworthy: Esteemed by its peers, Molpro isn’t just software; it’s an institution in the academic circles.
To put it simply, VASP doesn’t just sit in the background. It’s the silent force, the unsung hero in academic labs, driving the passion to decode the intricate dance of solid molecular structures.
Not merely a tool, but a confidant, helping researchers make sense of the enigmatic world of solid matter.
How much does it cost?
- Available on request
Source: https://www.vasp.at
#11. Molpro: Best for precision quantum chemistry
Summary
- High-precision focus
- Tailored for quantum chemistry
- Top-level molecular modeling programs
Molpro is less of a tool and more of a maestro when it comes to the dance of quantum chemistry. It doesn’t just compute; it crafts. For scholars venturing into the nuanced symphony of the quantum world, Molpro is akin to the seasoned conductor, directing each note with clarity, poise, and unparalleled precision.
Benefits
- High Precision: It’s not about mere accuracy; it’s about an impeccable, unwavering precision that Molpro delivers.
- Performance: Beyond just speed, Molpro offers an elegant efficiency in its computations, ensuring results are both fast and flawless.
- Trustworthy: Esteemed by its peers, Molpro isn’t just molecular modeling software; it’s an institution in the academic circles.
In the labyrinth of quantum chemistry where every detail matters, where every calculation holds profound implications, precision isn’t a luxury—it’s the very essence.
And that’s where Molpro doesn’t just shine—it dazzles, establishing itself as an indispensable companion for all serious quantum journeys.
How much does it cost?
- From $300/year
Source: https://www.molpro.net
#12. Autodock: Best for molecular docking simulations

Summary
- Specialist in docking simulations
- Detailed molecular insights
- Recognized in academia
Autodock isn’t just a program; it’s the seasoned captain steering the ship of molecular exploration. Picture a researcher, eager to chart the unexplored waters of molecular interactions, with Autodock as their trusted compass, pointing toward groundbreaking discoveries.
In academia’s bustling halls, where the magic of molecules can rewrite the story of drug discovery and bioengineering, Autodock is the shining lodestar.
Benefits
- Crystal Ball Capabilities: It doesn’t just show the present; it whispers the secrets of molecular interactions, predicting the unseen with finesse.
- Friendliest Navigator: Even amidst the stormy seas of complex simulations, this free molecular modeling software remains an intuitive ally, ensuring smooth sailing for researchers.
- All-in-One Toolkit: Whether it’s a gentle dock or a dynamic molecular dance, Autodock has the perfect tool for every nuance.
In the grand tapestry of molecular academia, where each thread represents an interaction waiting to be deciphered, Autodock isn’t just another needle—it’s the master weaver. With its unparalleled abilities, the enigmas of molecular docking transform into stories of precision and wonder.
How much does it cost?
- Free
Source: https://autodock.scripps.edu
#13. Discovery Studio: Best for life science molecule modeling
Summary
- Life sciences specialization
- Detailed molecular modeling
- Academically revered
Discovery Studio elevates life science research by offering tailored tools for molecular modeling. In a field where understanding the minutiae can lead to groundbreaking discoveries, this software ensures every molecular detail is accessible and comprehensible.
Benefits
- Tailored: Specifically designed for life sciences
- Depth: Ensures granular insights into molecules
- Collaborative: Promotes team-based academic research
The realm of life sciences demands precision, depth, and adaptability. Discovery Studio, with its meticulous molecular modeling capabilities, answers this call, solidifying its status as an academic linchpin.
How much does it cost?
- Free
Source: https://discover.3ds.com
#14. ChimeraX: Best for visual molecular analysis
Summary
- Premier visualization tools
- Focused on molecular analysis
- User-friendly interface
ChimeraX shines as a beacon for visual molecular analysis, transforming abstract data into vivid, comprehensible visuals. It’s not just about seeing—it’s about understanding, and ChimeraX ensures that the complex world of molecules is beautifully and clearly represented.
Benefits
- Visualization: Transforms data into insightful visuals
- Detail-Oriented: Captures every nuance of chemical structures
- Interactivity: Facilitates hands-on academic exploration
In the academic realm, where visual comprehension can bridge understanding, ChimeraX stands out. It seamlessly marries data with visual representation, becoming an indispensable tool for those seeking clarity in molecular analysis.
How much does it cost?
- Free
Source: https://www.cgl.ucsf.edu
#15. Materials Studio: Best for materials modeling
Summary
- Focused on material science
- Advanced modeling capabilities
- Highly recommended in academia
Materials Studio emerges as a front-runner when the quest is to unravel the complexities of materials at a molecular level. In academic circles, where breakthroughs in material science can reshape industries and technologies, this software provides a formidable platform for transformative research.
Benefits
- Comprehensive: Wide range of tools for varied material types
- User-Centric: Interfaces designed with academic needs in mind
- Advanced: Incorporates the latest in material modeling methodologies
When academia seeks to push the boundaries of material science, they often turn to Materials Studio. Its holistic approach, combined with cutting-edge tools, solidifies its place as an academic cornerstone in material research.
How much does it cost?
- $3000/year
Source: https://www.3ds.com
#16. Avogadro: Best for molecular structure editing
Summary
- Precision editing tools
- Molecular focus
- Favored by researchers
Avogadro stands out as the gold standard for molecular structure editing. Academic pursuits often require meticulous adjustments and insights into molecular structures, and Avogadro ensures that each tweak and transformation is executed with precision.
Benefits
- Flexibility: Adaptable to various molecular editing needs
- Precision: Fine-tuned tools for detailed editing
- Interactive: Intuitive UI for hands-on academic use
For academics, the devil often lies in the details. Avogadro, with its precision-driven toolset, ensures that researchers can dive deep into the intricacies of molecular structures, making it an indispensable asset for molecular research.
How much does it cost?
- Free
Source: https://avogadro.cc
#17. ROCS: Best for shape-based molecule alignment
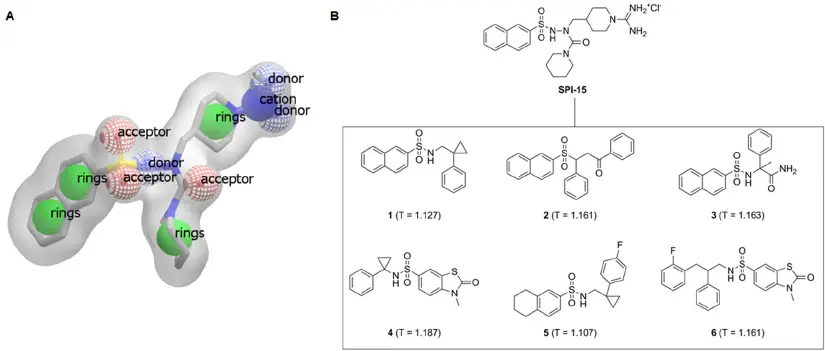
Summary
- Specialist in molecular alignment
- Uses shape-based techniques
- Highly regarded in academia
ROCS, with its focus on shape-based molecule alignment, offers a unique perspective into the world of molecular interactions. When academics aim to understand or predict how molecules align and interact based on their shape, ROCS stands as the go-to tool, shedding light on these critical alignments.
Benefits
- Unique Approach: Shape-based alignment offers distinct insights
- Precision: Ensures accurate alignment every time
- Compatibility: Integrates well with other molecular tools
Molecular alignment is pivotal in various research endeavors, and having a tool like ROCS, which emphasizes shape-based techniques, adds a layer of depth to academic pursuits. It’s not just about aligning; it’s about understanding the ‘why’ behind those alignments.
How much does it cost?
- Free
Source: https://www.eyesopen.com
#18. TINKER: Best for diverse molecular simulations
Summary
- Broad molecular simulation capabilities
- Versatile tools
- Acclaimed in the academic world
TINKER isn’t just another software—it’s the multi-tool pocket wonder of the molecular world! Picture a scientist standing at the edge of a vast molecular terrain, and TINKER is that trusted, versatile toolbelt, equipped for every twist and turn.
In academia, where the ebb and flow of research are as diverse as the landscapes of our planet, TINKER becomes the compass, guiding through the rich tapestry of molecular wonders.
Benefits
- A Spectrum of Tools: Imagine a color palette, but for molecular simulations. Whether it’s a pastel-hued study or a bold exploration, TINKER’s got the shade!
- Smooth Sailing: It’s tailored to fit academia’s glove, making it a breeze to navigate even the most complex molecular mazes.
- Detail-Oriented Explorer: No stone is left unturned. Every molecular nook and cranny is brought into the limelight with TINKER’s comprehensive approach.
In the buzzing hive of academic research, where the right tool can mean the difference between a breakthrough and a breakdown, TINKER doesn’t just participate—it takes the center stage.
Offering more than just solutions, it’s the trusty sidekick ensuring every molecular quest is both adventurous and enlightening.
How much does it cost?
- Free
Source: https://dasher.wustl.edu
#19. OpenMM: Best for high-performance molecular dynamics
Summary
- Specialized for molecular dynamics
- High-speed performance
- Preferred by leading academics
OpenMM isn’t merely software; think of it as a dynamic whirlwind, always raring to delve into the microscopic dance of molecules. In academic havens, where every tick of the simulation clock can unravel a universe of understanding, OpenMM is the torchbearer, casting light on the ever-evolving saga of molecular tales.
Benefits
- Flash and Dash: It’s like having a molecular superhero at your fingertips, zipping through computations with zeal.
- Pinpoint Precision: No guesswork here. It’s all about capturing the molecular ballet with grace and exactitude.
- Shape-Shifting Prowess: Whether you’re exploring a quiet molecular brook or a tumultuous atomic storm, OpenMM adapts, ready for every research challenge.
In the realm of academic exploration, where mediocrity just won’t do, OpenMM doesn’t merely make an appearance—it steals the show. Because it’s not just about the rush of speed, but the alchemy of accuracy and zeal that truly charts new frontiers in molecular dynamics research.
How much does it cost?
- Free
Source: https://openmm.org
#20. Schrodinger Suite: Best for comprehensive molecular research simulations
Summary
- Holistic suite of molecular tools
- Comprehensive simulations
- Esteemed in academia
Schrodinger Suite isn’t your average software; picture it as a thriving jungle of tools, where each instrument intertwines to form a vibrant web for molecular research.
In the halls of academia, where researchers seek to uncover the secret whispers of molecules, the Schrodinger Suite is their trusty guide, revealing the mystique of every molecular twist and turn.
Benefits
- All-in-One Treasure Trove: It’s like walking into a molecular bazaar – everything you need, right there under one starlit canopy.
- Spot-On Every Time: When it simulates, it doesn’t just play; it hits the bullseye, winning the trust of scholars everywhere.
- Harmonious Symphony: No jarring notes here. Every tool flows into the next, creating a seamless, melodious research waltz.
In the grand theater of molecular academia, Schrodinger Suite isn’t just another performer; it’s the maestro, orchestrating a flawless show. When the quest is for perfection and thoroughness, this suite doesn’t just participate—it leads to the ovation.
How much does it cost?
- Available on request
Source: https://www.schrodinger.com
Conclusion
The top molecular modeling software offers advanced features that streamline the discovery and development process. These programs connect scientific innovation processes, providing tools for molecular graphics, molecular dynamics simulations, and flexible high-quality rendering of molecular models.
From automated homology modeling servers that predict amino acid mutations to functionalities catering to materials science, such as energy calculations and simulating chemical reactions, these software solutions stand out in the realm of research.
They not only enhance our understanding of complex molecular interactions but also bolster the bridge between conceptual models and tangible innovations